



Figure 1
CJD and MRI: typical imaging findings in a sporadic form of Creutzfeldt-Jakob Disease
SectionNeuroradiology
Case TypeClinical Cases
Authors
Valentina Cardì, Lorenzo Motta, Marco Dugo, Paolo Cucchi, Giuseppe Carità, Andrea Saletti, Roberto Galeotti
Connected authors
76 years, female
Clinical History
A 76-year-old woman presented with a 2-month history of cognitive decline, unsteady gait, visual hallucinations, tremor on upper arms, aggressive behaviour.
The neurological examination revealed temporospatial disorientation, poor speech with mask-like face, hypokinesia, hyperexcitable t-reflex in upper arms, diffuse rigidity, hemisomatoagnosia.
Imaging Findings
On NE-CT, no significant anomalies were observed. It showed diffuse cerebral atrophy and some little ischemic injuries in basal ganglia regions. (Fig.1A-1B).
An MRI was performed, showing on T2w and FLAIR images enlargement sulci and basal cisterns indicating cerebral atrophy. (Fig.2A)
On FLAIR, bilateral and symmetrical cortical hyperintensities were seen in occipital lobes and in the right frontoparietal regions. (Fig.2B-2C).
On T1w, no significant anomalies were founded. (Fig.3A)
On T1w after intravenous contrast, no abnormal enhancement was seen in brain parenchyma, dura and leptomeninges. (Fig.3B)
On DWI/ADC, cortical diffuse signal restriction in occipital lobes and in the right frontoparietal regions (“ribbon sign”). (Fig.4A-4B)
Discussion
Creutzfeldt-Jakob disease (CJD) is a pathology belonging to a larger family of fatal neurodegenerative disorders known as transmissible spongiform encephalopathies. The incidence of CJD is about 1 – 1.5 cases per million per year [1].
CJD is caused by accumulation of misfolded prion proteins (prions) inside some brain cells, then spreading in all the cerebral tissues. It’s related to genetic mutation (Prp gene – codon 129) that causes uncontrolled prion collection inside cells.
Three forms of CJD are known: sporadic (sCDJ), familial and acquired.
The most frequent is the sCDJ, representing the 80-95% of the entire group: it occurs spontaneously between 55 to 70 years old and has a poor prognosis, with a 1-year mortality rate of 90% [2; 3]. It’s clinically characterized by rapidly progressive dementia and development of neurological symptoms.
The diagnosis is difficult: it’s based on clinical symptoms, pathognomonic EEG, typical MRI findings, and detection of protein 14-3-3 in the cerebrospinal fluid. The certain diagnosis is made by hystological sample obtained from biopsy or autopsy (accumulation of an abnormal form of the human prion protein PrPSc).
Today MRI is considered the most sensitive technique for the detection of CJD-related abnormalities in vivo. In fact, MRI showed cortical changes seen as hyperintensities in FLAIR and T2w-images in cortical regions, especially neocortical gyrus (the most common early manifestations), but also in basal ganglia and thalamus (“hockey stick sign”) [4].
Diffusion-weighted imaging (DWI) is probably the most sensitive and specific test for diagnosis of sCJD, showing persistent restricted diffusion (considered the most sensitive sign) as a ribbon-like signal hyperintensity of cerebral cortical gyri (“ribbon sign”).
Restricted diffusion can increase through the course of disease, but it could disappear in the last stages, maybe for atrophy of these regions [5].
In our case, EEG showed slow-waves abnormalities in frontotemporal regions with a global diffusion tendency.
CRL biochemical analysis showed an increase of TAU protein (1404 pg/ml, with a cut-off for suspected CJD >1250 pg/ml).
The research of 14-3-3 protein was positive; genetic tests confirmed codon 129 polymorphism of prion protein gene (PRNP).
Clinical history of rapid cognitive decline, extrapyramidal symptoms, suggestive EEG and cortical hyperintensities in DW-MRI and FLAIR-MRI, permit us to have diagnosis of CJD.
Differential Diagnosis List
Creutzfeldt-Jakob disease
Autoimmune encephalitis
Hypoxic Ischemic Encephalopathy
Infectious Disease (Encephalitis)
MELAS
Final Diagnosis
Creutzfeldt-Jakob disease
References
[1] Manix M, Kalakoti P, Henry M, et al. (2015) Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg Focus 39(5):E2. (PMID: 26646926)
[2] Alaoui A, Alami B, Habibi H, et al. (2019) Apport de l'IRM dans la maladie de Creutzfeldt Jakob: à propos d'un cas [MRI role in Creutzfeldt-Jakob disease: about a case]. Pan Afr Med J. 32:95. (PMID: 31223386)
[3] Bittar J, Joshi P, Genova J, et al. (2020) Creutzfeldt-Jakob Disease Presenting as Posterior Reversible Encephalopathy Syndrome. Cureus 12(3):e7211.
[4] Cammaroto, S, Smorto, C, Galletta D et al. (2015) Autopsy-like MRI findings: report on Creutzfeldt–Jakob disease in the end-stage. Neurol Sci 36:1497–1499. (PMID: 25690640)
[5] Geschwind MD, Potter CA, Sattavat M et al. Correlating DWI MRI With Pathologic and Other Features of Jakob-Creutzfeldt Disease. Alzheimer Dis Assoc Disord 23:82–87 (PMID: 19266702)
Case information
| URL: | https://eurorad.org/case/16916 |
| DOI: | 10.35100/eurorad/case.16916 |
| ISSN: | 1563-4086 |
License
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License.


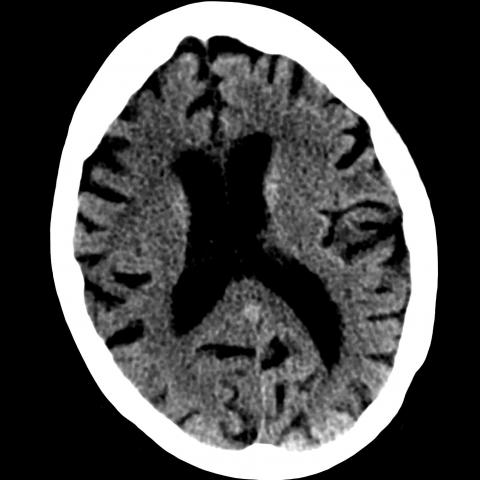
Figure 2

Axial MRI-T2w shows enlargement of sulci (yellow arrows) and basal cisterns indicating cerebral atrophy.

On sagittal and axial FLAIR, bilateral and symmetrical cortical hyperintensity in occipital lobes (red arrows).

On axial FLAIR cortical hyperintensity in the right fronto-parietal regions (red arrows).







Axial MRI-T2w shows enlargement of sulci (yellow arrows) and basal cisterns indicating cerebral atrophy.
© Department of Radiology, Sant’Anna Hospital, Ferrara, Italy
© Department of Radiology, Sant’Anna Hospital, Ferrara, Italy

On sagittal and axial FLAIR, bilateral and symmetrical cortical hyperintensity in occipital lobes (red arrows).
© Department of Radiology, Sant’Anna Hospital, Ferrara, Italy
© Department of Radiology, Sant’Anna Hospital, Ferrara, Italy

On axial FLAIR cortical hyperintensity in the right fronto-parietal regions (red arrows).
© Department of Radiology, Sant’Anna Hospital, Ferrara, Italy
© Department of Radiology, Sant’Anna Hospital, Ferrara, Italy



Axial DWI/ADC shows cortical diffuse signal restriction in occipital lobes (red arrows).
© Department of Radiology, Sant’Anna Hospital, Ferrara, Italy
© Department of Radiology, Sant’Anna Hospital, Ferrara, Italy

“Ribbon Sign”. Axial DWI/ADC shows cortical diffuse signal restriction in the right frontoparietal regions (red arrows).
© Department of Radiology, Sant’Anna Hospital, Ferrara, Italy
© Department of Radiology, Sant’Anna Hospital, Ferrara, Italy